Exploring Hund’s rule with high-performance computing
Some theories have stood strong for centuries and are taught in schools as part of the foundations of science. Naturally, these theories face their share of criticisms; such critiques often spark more debate, motivating new experiments. When a theory continues to hold up against challenges, it can rise to the status of a law. But has the Hund’s rule achieved this honour yet?
Hund’s rule, formulated by Friedrich Hund in 1925, is a set of widely accepted rules for predicting the ordering of the energy levels of atoms. The most important rule indicates that for a given electronic configuration, the state with the maximum number of electrons with unpaired spin is more stable (lower in energy) than a spin-paired state. This rule also guides how electrons fill degenerate orbitals (those with the same principal and angular quantum numbers, such as p, d, or f orbitals). Electrons occupy each orbital singly before pairing begins, which explains why half-filled and fully-filled orbitals are particularly stable due to their symmetry.
The stability of fully-filled (least spin-multiplicity) and half-filled subshells (maximum spin-multiplicity) arises from two main factors: symmetrical electron distribution and exchange energy. Symmetry leads to enhanced stability, as electrons in these subshells are distributed evenly, resulting in minimal shielding between them and allowing the nucleus to attract the electrons more strongly. Additionally, the exchange energy, which is a consequence of the indistinguishability of electrons, stabilizes electrons with parallel spins in degenerate orbitals. This exchange interaction is maximum when the subshell is half-filled. This concept forms the basis of Hund’s rule, where electrons occupy degenerate orbitals with parallel spins to maximize exchange interaction and stability.
For atoms like carbon, Hund’s rule correctly predicts that the triplet state (with unpaired electrons) is more stable than the singlet state (with paired electrons). For organic molecules, with typically a singlet ground state (S0), the rule suggests that the first excited triplet state (T1) is lower in energy than the first excited singlet state (S1). Hence, the singlet-triplet gap (energy of T1 subtracted from energy of S1) is usually positive for organic molecules. Recent computational studies have suggested that certain molecules, such as nitrogen-substituted triangulenes, might show a negative singlet-triplet gap, attracting significant attention from the scientific community. Experiments have indicated the likelihood of a negative gap for two such triangulenes. This, in turn, has consequences in designing efficient light-emitting molecules used in optoelectronic devices such as smartphone screens. Our research group focuses on data-driven discoveries. With access to high-performance computing resources, we analyzed 12,880 small organic molecules to identify any molecule challenging Hund’s rule (i.e., with a negative S1-T1 gap). Our advanced computational tools and Big Data techniques allowed us to investigate each molecule’s excited state energy trends. Our study identified several molecules as “false positives” in Hund’s rule violation while using an approximate quantum chemistry method and obeying the rule (i.e., “true negatives”) when modeled with a more accurate method. However, the quest to develop a computational method that can accurately account for exchange and correlation effects continues. If such a method predicts a negative singlet-triplet gap, it could reveal a “true positive” exception to Hund’s rule.
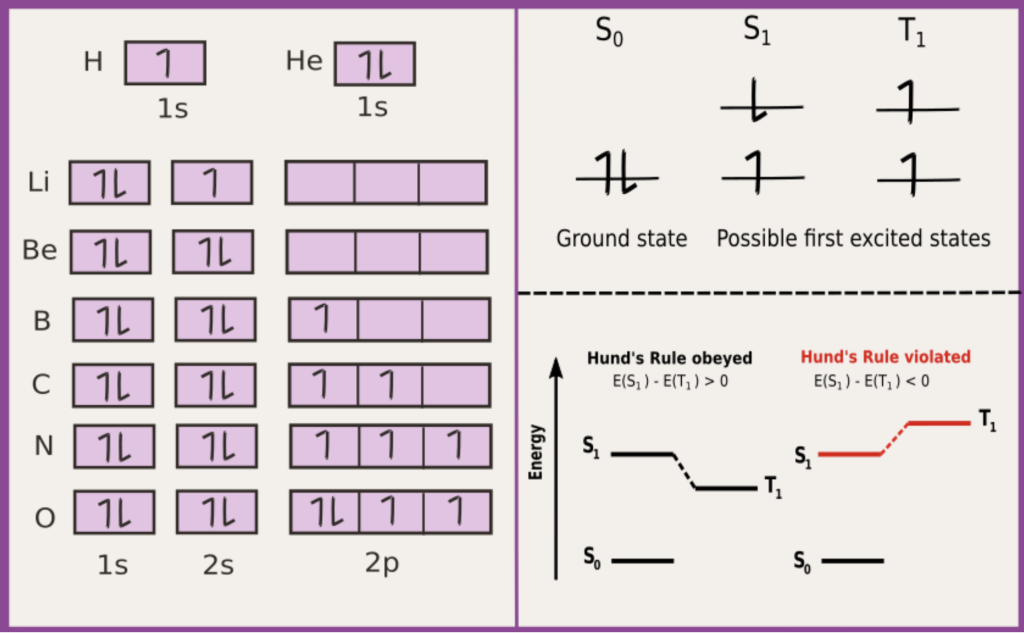
Fig caption: Electronic configurations of the ground state of different atoms (shown on the left) and different electronic states of an organic molecule (shown on the right). S and T denote singlet and triplet spin arrangements, respectively.
We presented all our findings as a Python module called pymoldis. This tool allows us to quickly check the excited state properties of any molecule in our dataset. Using pymoldis, we focused on ten molecules with the lowest singlet-triplet gaps. Most nitrogen-containing molecules showed something unusual—these molecules contained a sp3-nitrogen center with a planar bonding environment deviating from the ideal pyramidal geometry as in the ammonia molecule. We faced two challenges in this study: 1) identifying an accurate computational method for modeling singlet-triplet gaps and 2) automating the computational modeling to minimize manual data handling, which often introduces mistakes.
Looking ahead, we plan to build on the findings of our study, titled“Resilience of Hund’s Rule in the Chemical Space of Small Organic Molecules” (Accessible at https://doi.org/10.1039/D4CP00886C), which demonstrated that none of the molecules in this vast chemical space of small molecules violate Hund’s rule. Next, we will focus on larger, experimentally-relevant systems, such as azaphenalenes, which have been previously suggested to violate the rule.
The exploration of Hund’s rule continues, but the real question is how many molecules can break the rule—and whether they are genuinely breaking it. If so, what is the physical picture? These questions motivate our future research.
Author: Atreyee Majumdar (PhD student, Raghunathan Ramakrishnan’s research group, TIFR Hyderabad)
Target Audience: Students pursuing advanced mathematics, physics, chemistry and computer science.